
Predykcja struktury trzeciorzędowej białek
Deep learning do predykcji struktury białek
Predykcja struktury białka 3D na podstawie sekwencji aminokwasów.
Opis projektu
Celem projektu było przewidywanie kątów tworzących główną strukturę (szkielet) białka na podstawie sekwencji aminokwasów.
Przegląd
Predykcja struktury trzeciorzędowej białek to jedno z najważniejszych nierozwiązanych zagadnień biochemii. Obecne metody są kosztowne i niedokładne. Uczenie maszynowe daje szansę na tańsze i skuteczniejsze rozwiązania. Przełomem była praca Mohammeda AlQuraishiego End-to-end differentiable learning of protein structure, która wraz z RaptorX-Angle zainspirowała ten projekt.
Model powstał na potrzeby kursu, ale wyniki wykraczały poza standardowy zakres zajęć.
Szczegóły techniczne
Korzystamy z zestawu ProteinNet oraz piszemy cały pipeline przetwarzania danych w Tensorflow. Rdzeniem modelu jest CNN podobny do tego z RaptorX. Model trenujemy optymalizatorem ADAM na stratach MSE i MAE pomiędzy przewidywanymi i prawdziwymi kątami torsyjnymi.
LSTM z diagramu poniżej zastępujemy CNN i nie implementujemy różowej części, która zamieniałaby kąty na przestrzeń euklidesową 3D.
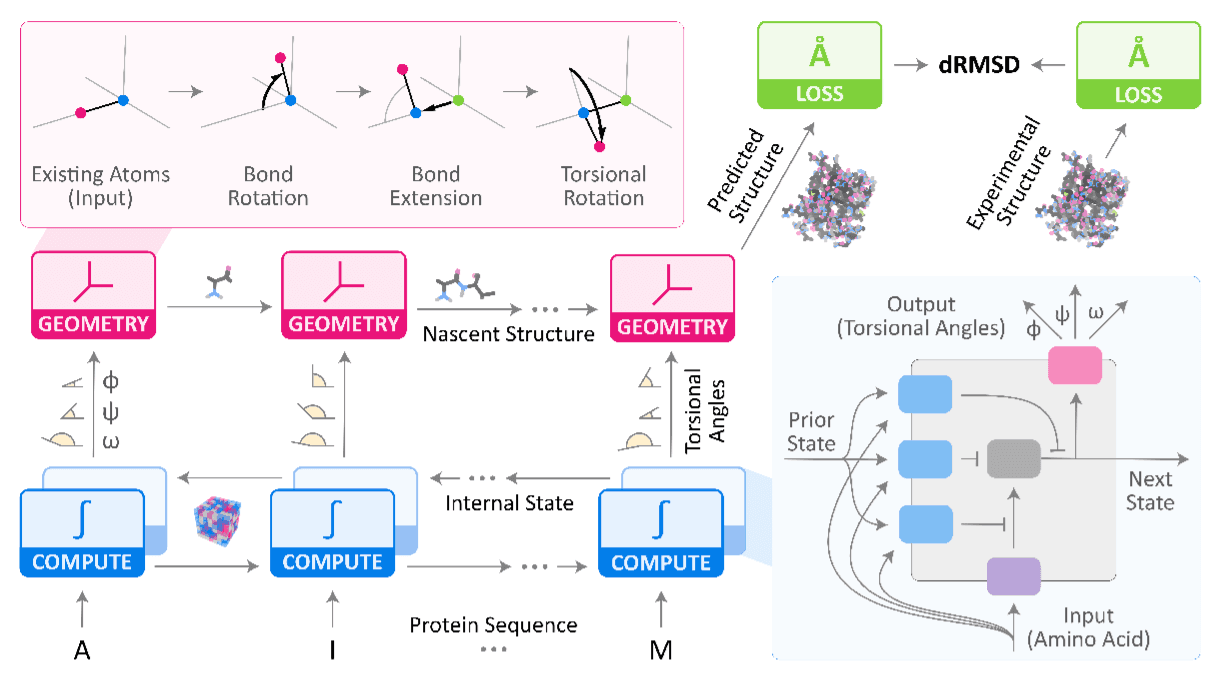
Główne wyzwanie to 1) wydajna funkcja straty oraz 2) kątowy charakter wyjścia sieci.
- W pracy AlQuraishiego struktura trzeciorzędowa jest aproksymowana przez 3 kąty torsyjne na aminokwas, a następnie odtwarzana w 3D do obliczenia straty. To kosztowne obliczeniowo, więc minimalizujemy stratę bezpośrednio na kątach, jak w pracy RaptorX.
- Wyjście sieci musi być zmapowane na zakres kątów. Jedna metoda to bezpośrednie przewidywanie 3 wartości ograniczonych do [-pi, pi] przez skalowany tanh. Inna to przewidywanie 6 wartości tworzących 3 pary wektorów 2D, które następnie zamieniamy na kąty funkcją atan2.
Wyniki
Model osiągnął wyniki porównywalne z RaptorX przy mniejszej przestrzeni cech. Raport końcowy dostępny jest tutaj.
Mój wkład
Wykorzystałem Tensorflow do wczytywania danych, przygotowałem pipeline kolejkowy i w pełni różniczkowalny graf, który konwertuje współrzędne euklidesowe atomów białka do kątów torsyjnych i minimalizuje stratę między przewidywaniami a prawdziwymi kątami.
Testowałem różne sposoby „angularizacji” wyjścia modelu i przygotowałem modularny kod dostępny na GitHubie.